Simulation data for tutorial
Reference genome
The first 500kb of chromosome 1 from the hg38 assembly was used as the reference genome. The extracted sequence was saved as chr1 in the FASTA format, which was then used for subsequent simulations and as reference genome for the tutorial.
Insertion sequence
The insertion sequence for the simulation was based on the pSLCAR-CD19-CD3z vector construct, obtained from Addgene (Plasmid # 135993).
The construct encapsulates a a CAR T cell vector sequence and was developed by Scott McComb and colleagues. The vector architecture is illustrated below with some key features highlighted, as well as with an illustration of the 100bp-long fragmentation used in the tutorial.
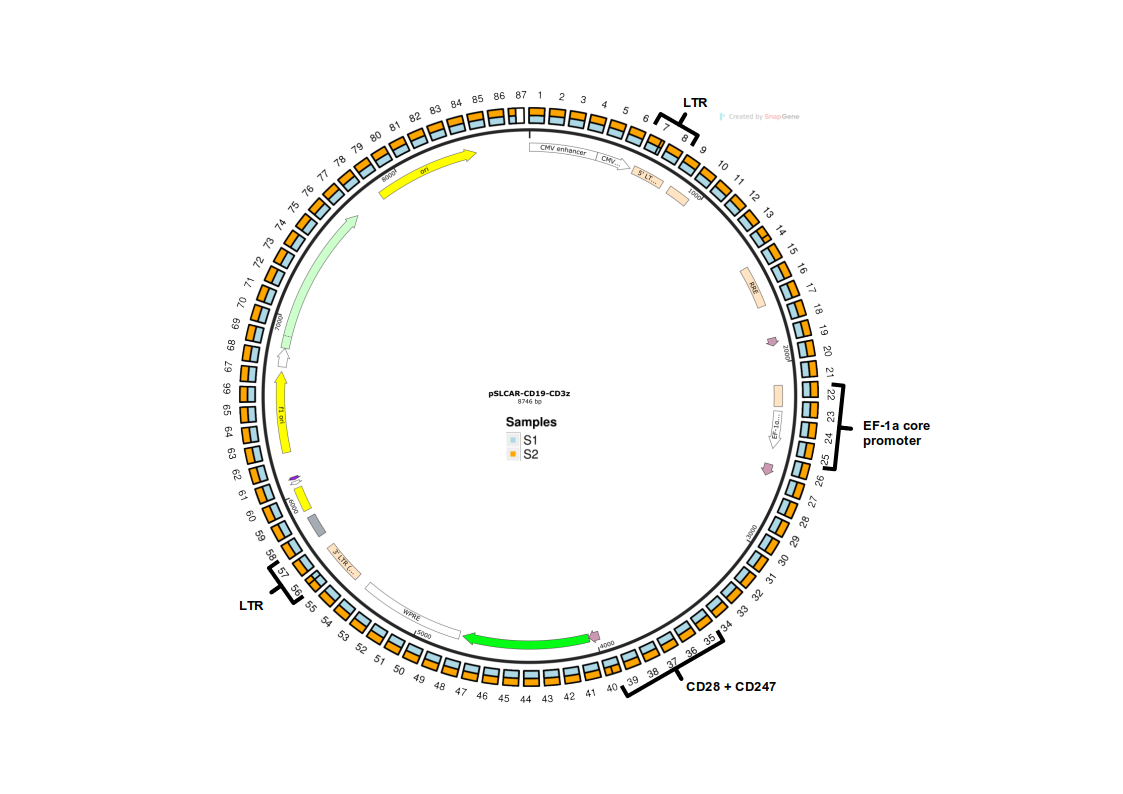
Further Details
Lentivirus-based transduction systems generally integrate the DNA sequence between the two long terminal repeats (LTRs). In a biological context, we would typically observe only the sequence between the 5' LTR and the 3' LTR, rather than the entire vector construct. To maintain simplicity in the simulated data, this specific adjustment to the vector sequence has not been made.
Read data generation
- Reference Genome Sampling: 1000 reads were sampled from the reference genome with a mean read length of 10,000 bp, generated from a log-normal distribution.
- Insertion Introduction: 5 of the 1000 reads were randomly chosen to receive an insertion, either the full-length or part of the construct, with random insertion directionality (
+or-). - Generation of Samples: The process was repeated twice to generate two samples (
S1andS2), each with a summary of the expected insertion locations. - BAM generation: Both samples were mapped against the reference genome to generate BAM files using:
minimap2 -ax map-ont chr1_1_50000_ref.fa S1.fa | samtools sort | samtools view -F 2304 -o S1.bam
This simulation provides a set of reads with varying insertion locations for evaluating the pipeline's detection and analysis capabilities.
Full Simulation Code:
#!/usr/bin/env python3
import random
import numpy as np
from Bio import SeqIO
from Bio.Seq import Seq # For reverse complement
# Paths to input files
reference_genome_path = "chr1_1_50000_ref.fa" # Reference genome
vector_sequence_path = "vectorseq.fa" # Vector sequence
random.seed(2101) # Seed for random, S1: 2101 and S2: 2015
np.random.seed(2101) # Seed for numpy.random, S1: 2101 and S2: 2015
def collapse_fasta(fastapath):
"""Collapses multi-line FASTA sequences into a single string."""
with open(fastapath, 'r') as fasta_file:
seq_list = []
for record in SeqIO.parse(fasta_file, 'fasta'):
seq_list.append(str(record.seq))
return ''.join(seq_list)
def generate_reads(reference_genome, mean_read_length=10000, num_reads=1000):
"""Generates random reads from the reference genome."""
reads = []
# Create read lengths following a log-normal distribution
read_length_distribution = np.random.lognormal(mean=np.log(mean_read_length) - 0.5, sigma=1.0, size=num_reads)
for read_length in read_length_distribution:
read_length = int(read_length) # Ensure lengths are integers
start_position = random.randint(0, len(reference_genome) - read_length)
read = reference_genome[start_position:start_position + read_length]
reads.append(read)
return reads
def add_insertions_to_reads(reads, insertion_sequence, num_insertions):
"""Adds insertions with strandedness to randomly selected reads."""
insertion_summary = [] # To store insertion details for the summary
for _ in range(num_insertions):
strand = None
# Randomly select a read to modify
read_index = random.randint(0, len(reads) - 1)
read = reads[read_index]
# Decide whether to use the full insertion or a partial sequence
if random.choice([True, False]): # 50% chance
insertion = insertion_sequence # Full insertion
else:
start = random.randint(0, len(insertion_sequence) - 2000)
end = start + random.randint(2000, min(5000, len(insertion_sequence) - start))
insertion = insertion_sequence[start:end] # Partial insertion
# Decide the strandedness of the insertion
if random.choice([True, False]): # 50% chance for negative strand
insertion = str(Seq(insertion).reverse_complement()) # Reverse complement for negative strand
strand = "-"
if strand is None: #default to positive
strand = '+'
# Insert the sequence at a random position in the read
insert_position = random.randint(0, len(read))
modified_read = read[:insert_position] + insertion + read[insert_position:]
# Replace the original read with the modified read
reads[read_index] = modified_read
# Store the details of the insertion
insertion_summary.append((f'Read-{read_index + 1}', len(insertion), strand))
return reads, insertion_summary
# Collapse the FASTA files into single sequences
reference_genome = collapse_fasta(reference_genome_path)
vector_sequence = collapse_fasta(vector_sequence_path)
# Generate reads and add insertions
non_insertion_reads = generate_reads(reference_genome) # Reads without insertions
insertion_reads, insertion_summary = add_insertions_to_reads(non_insertion_reads.copy(), vector_sequence, 5)
# Combine reads and save them to a file
total_reads = insertion_reads + non_insertion_reads
output_file_path = "S1.fa"
with open(output_file_path, 'w') as output_file:
for i, read in enumerate(total_reads):
output_file.write(f'>Read-{i+1}\n{read}\n')
# Print the summary of insertions
print("Summary of Inserted Reads:")
for read_name, insertion_length, strand in insertion_summary:
print(f"{read_name}: Insertion length = {insertion_length}, Strand = {strand}")
# Save summary to a file
summary_file_path = "S1_InsertionSummary.txt"
with open(summary_file_path, 'w') as summary_file:
summary_file.write("Summary of Inserted Reads:\n")
for read_name, insertion_length, strand in insertion_summary:
summary_file.write(f"{read_name}: Insertion length = {insertion_length}, Strand = {strand}\n")
# Debugging outputs
print(f"Total reads: {len(total_reads)}")
Summary of Inserted Reads for S1:
Read-745: Insertion length = 8746, Strand = -
Read-555: Insertion length = 8746, Strand = +
Read-561: Insertion length = 2248, Strand = +
Read-343: Insertion length = 8746, Strand = +
Read-902: Insertion length = 8746, Strand = +
Summary of Inserted Reads for S2:
Read-522: Insertion length = 8746, Strand = +
Read-824: Insertion length = 8746, Strand = -
Read-262: Insertion length = 2532, Strand = -
Read-417: Insertion length = 2593, Strand = -
Read-682: Insertion length = 8746, Strand = +
Annotation data processing
The gene annotation data was obtained from UCSC by selecting the region chr1:1-500,000 from the All GENCODE V44 track and downloading the selection with the "All fields from selected table" output format.
Full Annotation Data Processing
The raw donloaded table had the following format:
#bin name chrom strand txStart txEnd cdsStart cdsEnd exonCount exonStarts exonEnds score name2 cdsStartStat cdsEndStat exonFrames
585 ENST00000456328.2 chr1 + 11868 14409 11868 11868 3 11868,12612,13220, 12227,12721,14409, 0 DDX11L2 none none -1,-1,-1,
585 ENST00000619216.1 chr1 - 17368 17436 17368 17368 1 17368, 17436, 0 MIR6859-1 none none -1,
585 ENST00000473358.1 chr1 + 29553 31097 29553 29553 3 29553,30563,30975, 30039,30667,31097, 0 MIR1302-2HG none none -1,-1,-1,
585 ENST00000469289.1 chr1 + 30266 31109 30266 30266 2 30266,30975, 30667,31109, 0 MIR1302-2HG none none -1,-1,
...
cat UCSC_genes_chr1_0_500000 | cut -f 3,4,5,6,13 | awk -v OFS="\t" 'NR > 1 {print $1, $3, $4, $5, ".", $2}' - | sort -k1,1 -k2,2n > UCSC_genes_chr1_0_500000
chr1 11868 14409 DDX11L2 . +
chr1 17368 17436 MIR6859-1 . -
chr1 29553 31097 MIR1302-2HG . +
...
show the vector sequence and how it is fragmented into 100bp sized-fragments